Cystic Fibrosis
Cystic fibrosis is an inherited condition in which glands produce excessively sticky mucus. The sticky material clogs the lungs, liver, pancreas, and intestines and makes it difficult to breathe and to digest food properly.
KEYWORDS
for searching the Internet and other reference sources
Chest physical therapy
Gene therapy
Mucoviscidosis
Phenylalanine
Pulmonary system
Rachel's Story
Rachel's parents were worried. They believed they provided all the proper care Rachel needed, just as they had for their older daughter when she was a baby. But Rachel failed to gain as much weight as other children her age, even though she seemed to have the typical appetite of an infant. Rachel also seemed to have more colds than other children had and a lot more colds than her older sister ever had. She coughed often and breathed with a wheezing sound. Then, around the time of her second birthday, Rachel developed pneumonia * .
Rachel's failure to gain weight and her frequent respiratory infections led her doctor to suspect that Rachel had cystic fibrosis (SIS-tik fy-BRO-sis), which is usually known by its initials, CE Tests confirmed the diagnosis.
* pneumonia is an inflammation of the lungs, usually caused by bacteria, viruses, or chemical irritants.
CF is a hereditary disease that affects about 30,000 children and adults in the United States. It is the most common hereditary condition that affects people of European ancestry, occurring in 1 of every 2,000
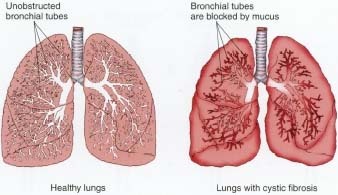
What Is Cystic Fibrosis?
Cystic fibrosis is a chronic, hereditary disease that affects many of the body's organ systems. In CF, some of the thin, easy-flowing mucus * in the body's respiratory and digestive systems become thicker. Glands * in the body produce mucus to do such things as lubricate the lungs, trap dust and bacteria that is inhaled through the nose, and protect the lining of the intestines from the acidic fluids that help digest food.
The glands in people with CF, however, produce sticky mucus that clogs the passageways in the lungs, which makes it difficult to breathe and leads to infections. The sticky mucus also blocks the easy flow of digestive acids and enzymes from the pancreas * and liver * to the intestines. Without adequate amounts of these digestive fluids in the intestines, people with CF cannot break down their food into the substances the body needs for nourishment.
* mucus is a combination of water, salt, cells, and other material that forms a coating that lines the respiratory and digestive systems.
* glands are groups of cells that act on substances in the bloodstream to change them for use in other parts of the body or to remove them from the body.
* pancreas is an organ in the upper abdomen that secretes enzymes to help with digestion.
* liver is a large organ with many functions, including secreting the digestive fluid bile.
* genes are chemicals in the body that help determine a person's characteristics, such as hair or eye color. They are inherited from a person's parents and are contained in the chromosomes found in the cells of the body.
* amino acids are the chief components of proteins.
What Causes Cystic Fibrosis?
CF is caused by a mutation in a gene * on chromosome 7. Chromosome 7 is one of 23 pairs of chromosomes that are part of each person's genetic makeup. The CF gene causes the production of a protein that lacks an important amino acid * , phenylalanine (fen-il-AL-a-neen). Without that amino acid, the protein hinders the ability of mucus to obtain the proper amounts of water and salt from the body, which the mucus needs to maintain its thin and easy-flowing texture. This turns the mucus into a dense, sticky substance that clogs the respiratory and digestive systems.
The obstructions in the lungs make it hard to breathe and can increase the risk of infections. The problems in the digestive system prevent the body from getting all the nutrients it needs from food. It also means people
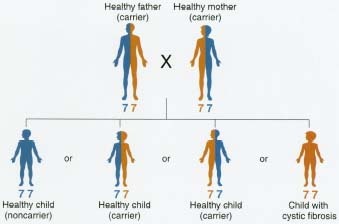
A person may carry the CF gene on one of the two copies of chromosome 7 and not have any signs of CE This person is called a carrier * . Parents can pass the CF gene to many generations of offspring. When a person has the CF gene on both copies of chromosome 7, then that person will have CE When parents are both carriers, such as Rachel's parents, their children have a one in four chance of having CE Estimates are that 10 million Americans, or 1 out of 29, carry the CF gene.
How Do Doctors Know Someone Has CF?
As far back as the 1600s, there were descriptions of children with symptoms of cystic fibrosis. It was not until 1938 that it was recognized as a separate disease, because lung infections are common to many conditions. Even today, the symptoms of CF sometimes can be confused with pneumonia or asthma.
* carriers are people who have the genes for a disease without having the disease itself.
Symptoms
Cystic fibrosis affects each person differently. Many people with CF do not appear to be severely ill. In general, people with CF have some or all of these symptoms:

- Salty-tasting skin and sweat
- Persistent cough or wheezing
- Many respiratory infections
- Bulky, smelly stools or bowel movements
- Nasal polyps (small growths in the nose)
- Enlargement of the fingertips and toes (clubbing)
Also, people with CF may eat large amounts of food but still be hungry. The food is not being digested properly, because the mucus is blocking the ability of digestive enzymes * and acids to break down food and absorb nutrients. Large portions of the poorly digested food are passed out during bowel movements. The individual may lose weight.
Later the pancreas may clog up and fail to secrete the enzymes essential to normal digestion. The liver becomes clogged, which may lead to cirrhosis (si-RO-sis), a condition in which the liver becomes hardened and fails. Diabetes also develops frequently in people with CF as they get older.
Diagnosis
Doctors may begin to suspect CF soon after birth when the baby becomes ill with repeated respiratory infections, fails to gain weight despite a healthy appetite, and shows other symptoms of CE In addition, about 10 percent of infants with CF have intestinal blockage due to thick mucus that is apparent at birth. Many of the symptoms of CF are common in people who do not have the disease, but there are tests to confirm a person has CF.
A sweat test is considered the best method to diagnose cystic fibrosis because it is relatively easy to perform and is accurate. The test determines the salt content of perspiration. Although sweat can seem salty in people without CF, the level of salt in the perspiration of people with CF is higher. A more complicated test looks for the CF gene on both copies of chromosome 7.
A Morning Ritual for Rachel
After her doctor determined that Rachel had cystic fibrosis, her parents worried she would not have a chance to do many of the same things as her older sister or other children. But as she grew, Rachel went to school, participated in sports, and did many other everyday activities.

Most people with CF get treatment that involves helping them breathe more easily and digest food better, which makes day-today activities less difficult. Although their symptoms can range from mild to severe, many people with CF receive treatments similar to Rachel's.
For example, Rachel receives chest physical therapy. Each morning, her parents vigorously thump Rachel's back and chest to help loosen the thick mucus in her lungs so she can cough it out. They learned the technique from a physical therapist after Rachel was diagnosed with CF.
* enzymes are natural substances that speed up specific chemical reactions in the body.
Coughing is one of the main ways that people with CF can clear the mucus from their lungs. In school, Rachel's teachers were told to expect her to cough often. Although the teacher and the students knew Rachel had CF, they did not make a big deal about her coughing. Rachel kept her own box of tissues on her desk, so she could cough the mucus into it and toss it into a nearby garbage can.
Rachel also participated in physical education classes. Exercise is another way that people with CF loosen the mucus in their lungs. Sometimes Rachel got tired more quickly than the other children, because she could not breathe as easily. But she joined in many of the exercises and games on most days.
At lunch and other times she ate a meal or snacks, Rachel took pills. The pills contained enzymes to help her digest food. Without them, the mucus in her digestive system would prevent her from getting the nutrients she needs from food. Even with the enzyme pills, Rachel and others with CF often need to take vitamin supplements and eat a diet rich in nutrients to assure they get the proper nourishment.
Rachel and others with CF also take antibiotics to prevent or treat lung infections. Sometimes, the antibiotics are taken as pills or inhaled into the lungs using a device called a nebulizer. People with CF also sometimes take prescription medications that thin the mucus and help reduce lung inflammation, which makes breathing easier and helps reduce the number of lung infections.
Perhaps the most exciting news for people with CF like Rachel was the discovery of the CF gene in 1989. It has led to research into gene therapy.
Living with CF
Once, CF almost always caused death in childhood. But treatments in recent decades have allowed many people with cystic fibrosis to live into adulthood. As with children, the symptoms for an adult can range from mild to severe, but eventually the recurring infections in the lungs begin to damage the lung's ability to function. This is the usual reason people with CF eventually die.
The Cystic Fibrosis Foundation says that now the average life expectancy of people with CF is 31 years. That is many years longer than in the past, but it still means only half of the people with CF will live that long. About half will live longer. With treatment, people with CF are able to do many of the things that other people do. And with work continuing in gene therapy, there is optimism that CF research is advancing toward a cure.
Turning on the CF Gene
In 1989, scientists discovered the location of the CF gene. If they could replace the defective gene with a normal gene, then they would be able to cure the cells that produce the defective CF protein. This would mean the mucus in the respiratory and digestive systems would be thin and easy flowing as opposed to thick and sticky.
In 1990, two teams of researchers were able to correct CF cells in lab dishes, by adding normal copies of the gene. In the spring of 1993 the first experimental dose was given to a person with CF. In October 1993, scientists determined that gene treatment had repaired a damaged gene in a human patient.
Gene therapy is a complicated experimental process. Scientists are beginning to understand what methods can be used to deliver the gene to the parts of the body where it can do its work. And they are studying how often the treatment would need to be repeated to assure the best results.
It could be that the gene therapy never would be able fix all of the defective genes, but it might cause enough of them to work properly to improve the quality of life for people with CF.
Boomer Fights Back
When Gunnar Esaison was diagnosed in 1993 with cystic fibrosis, his father, Boomer Esaison, decided to fight back. The Cincinnati Bengals quarterback, National Football League star, and television sports reporter had been a fierce competitor for many years. Now, his greatest battle became the fight against CF.
The NFL star started the Boomer Esaison Foundation, with the main goal being to find a cure for CF. The Foundation supports basic research and clinical trials for new CF treatments.
Resources
Books
Grinshaw, Joshua. My Heart Is Full of Wishes. Austin, TX: Raintree-Steck-Vaughn, 1995. A young boy with CF describes his dreams.
Harris, Ann. Cystic Fibrosis: The Facts. New York: Oxford University Press, 1995.
Silverstein, Alvin. Cystic Fibrosis. New York: Venture Books, 1994.
Organization
Cystic Fibrosis Foundation, 6931 Arlington Rd., Bethesda, MD 20814
http://www.cff.org
See also
Asthma
Cirrhosis
Genetic Diseases
Pneumonia
Comment about this article, ask questions, or add new information about this topic: