Sickle-cell Anemia
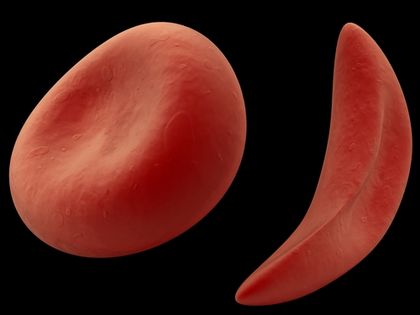
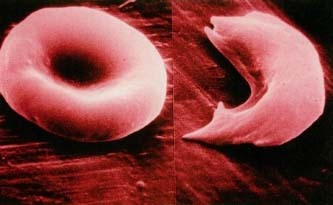
Sickle-cell anemia, also called sickle-cell disease, is a hereditary dis order in which abnormal hemoglobin * within the red blood cells (RBCs) causes the cells to take on abnormal sickle (crescent) shapes. This decreases the ability of the hemoglobin to transport oxygen throughout the body. The sickled cells tend to bunch up and clog the blood vessels, and they tend to break apart more easily than normal RBCs. This may cause inflammation, pain, tissue damage, and anemia
KEYWORD
for searching the Internet and other reference sources
Inherited diseases
What Are the Sickle-Cell Trait and Sickle-Cell Disease?
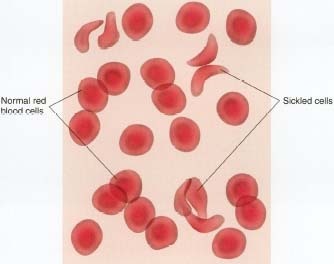
Normally, red blood cells (RBCs) are rounded and flattish, like a saucer. They pass easily through the tiniest blood vessels. Red blood cells sickle or take on the crescent shape when they carry an abnormal form of hemoglobin called hemoglobin S. The abnormality in hemoglobin S occurs because of the presence of a faulty gene * .
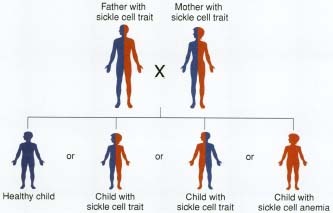
People inherit one set of genes from each parent. They can inherit either two normal hemoglobin genes (HbA), one normal hemoglobin gene and one gene for the abnormal hemoglobin gene (HbS), or two abnormal HbS genes, depending on the composition of their parents' genes.
* hemoglobin (HEE-mo-globin) is a protein found in red cells. It binds to oxygen so that oxygen can be carried by way of the circulatory system throughout the body for use by other cells.
* anemia (a-NEE-mee-a) is a condition in which the amount of oxygen transported by the blood is reduced either because of a reduced number of RBCs or because there is too little hemoglobin in the RBCs.
* genes are chemicals inherited from both parents that help determine physical characteristics, such as whether a person has brown hair or blue eyes, or in this case, normal or abnormal hemoglobin.
When a person carries one HbS and one HbA gene, the presence of the normal gene is sufficient to override the effects of the HbS gene so
When people inherit two abnormal HbS genes, one from each parent, they have sickle-cell disease, and they show symptoms of that disease. They have only abnormal HbS genes to pass on to their children.
Since sickle-cell disease is a genetic disorder, its frequency varies in different populations worldwide. It is found most frequently in Africa, where in some locations up to 40 percent of the population has at least one HbS gene. The gene is also found in people in Mediterranean and Middle Eastern countries, such as Italy, Greece, and Saudi Arabia. There are groups of people in India, Latin America, the Caribbean, and the United States where the HbS gene is also found.
Among people of African ancestry in the United States, about 8 in every 100 individuals carries at least one HbS gene (has the sickle-cell trait) and about 40,000 people carry two copies of the HbS gene and have sickle-cell disease.
* antibiotics (an-ty-by-OT-iks) are drugs that fight bacterial infection.
What Are the Effects of Sickle-Cell Disease?
People who have sickle-cell disease get infections more frequently than other people. In 1987 the National Institutes of Health recommended that all babies, regardless of ethnic background, be tested for the presence of the HbS at birth. Babies with sickle-cell disease are often given antibiotics * to prevent infections. Before this screening became common,
Infants, older children, and adults with sickle-cell disease also periodically experience bouts of critical illness called crises. They also suffer from complications of anemia.
Because sickle-cell disease is hereditary, people are born with it and will have it all their lives. There is no way one person can catch sickle-cell disease or sickle-cell trait from another, and there is no way the disease can be cured.
Crises
Sickle-shaped red blood cells clump more easily than normal RBCs. Sickle-cell crises start suddenly when clumping of sickled RBCs in the blood vessels obstructs the normal flow of blood, depriving various tissues and organs of oxygen. The first crises usually appear in early childhood.
Crises may be brought on by respiratory infection, by a loss of body fluids from vomiting or diarrhea, by situations where the body's need for oxygen is increased, or they may occur for no obvious reason. They may last for several days, and cause fever and sharp, intense pain in the back, abdomen, chest, arms and legs. In infants, the hands and feet may become swollen and painful.
Crises may damage nearly any part of the body, but especially the bones, kidneys, intestines, lungs, liver, spleen, and the central nervous system * , including the brain. There may also be eye damage, stroke, convulsions, or paralysis. The damage is caused because the clumping of RBCs in a blood vessel deprives tissues of oxygen.
Many people with sickle-cell disease go for long periods during which they may feel relatively well and engage in most normal activities and are free of crises (in remission). Others may experience pain on a daily basis, and some need to be hospitalized as a result of crises several times a year.
Anemia
Sickle cells are more easily broken down and destroyed than normal RBCs. Sickle cells have a lifespan of 10 to 20 days compared to 90 to 120 days for a normal RBC. People with sickle-cell anemia thus cannot keep up a normal level of oxygen-carrying hemoglobin in their blood. This is so despite the fact that they make red blood cells faster than people without the disease. The result is that they are anemic.
The anemia may become so severe that a person will need to have a blood transfusion. The long-term effect of anemia is that the heart has to work harder to pump more blood through the body. Over time the heart enlarges, increasing the risk of heart attack and heart failure.
Other complications of sickle-cell disease
People who have sickle-cell disease are more susceptible to all kinds of bacterial and fungal infections. They are more likely to have strokes * and to experience kidney failure. In some people, the liver enlarges, and by age 30, 70 percent of people with sickle-cell disease have developed gallstones.
Modern medicine has increased the survival of people with sickle-cell disease. About half the people with sickle-cell disease live beyond 50 years. Still, living with the pain and complications of this condition can cause emotional stress on both the person with the disease and the family.
How Is Sickle-Cell Disease Diagnosed?
Sickle-cell disease and sickle-cell trait can be diagnosed by a test that is much simpler than its name: hemoglobin electrophoresis (e-lek-tro-fo-REE-sis). This test detects the presence of HbS and other abnormal hemoglobins. A complete blood count (CBC) counts the number of RBCs and checks for abnormal shapes.
* central nervous system refers to the brain and the spinal cord, which coordinate the activity of the entire nervous system.
* stroke is a blocked or ruptured blood vessel within the brain, which deprives some brain cells of oxygen and thereby kills or damages these cells. Also called apoplexy (AP-o-plek-see).
* dehydration (dee-hy-DRAY-shun) is loss of fluid from the body.
How Is Sickle-Cell Disease Treated?
There is no cure for sickle-cell anemia. Much treatment is preventive and directed toward symptoms. Antibiotics may be given to prevent infections. Fluid intake is important to prevent dehydration * , a major cause of sickling. Folic acid may be given daily to lessen the anemia by helping to make new red cells. Children are given a complete set of immunizations. Lifestyle habits that can help sickle-cell patients stay healthy and have fewer crises include drinking plenty of water, avoiding extremes of heat and cold, avoiding stress and overexertion, getting enough sleep, and having regular medical check-ups.
Treatment in a sickle-cell crisis may require oxygen therapy, pain relieving medications, antibiotics, and intravenous fluids to offset dehydration. Blood transfusions may also have to be performed. Treatment of pain is a major concern for physicians whose patients have sickle-cell disease. The benefits of different pain relievers and their unwanted side effects must be balanced for each patient.
Research is ongoing to find better ways to treat people with sickle-cell disease. Some of these research efforts are directed at stimulating the production of fetal hemoglobin, a form of hemoglobin found in infants, even those with sickle-cell disease. Other research is directed toward the development of drugs that block dehydration in cells. Gene therapy and the transplantation of healthy bone marrow that makes normal red blood cells are also under investigation.
A Possible Benefit of
the Sickle-Cell Trait
Sickle-cell disease is especially prevalent among people of African or African American descent. No one knows for sure why this is so. However, it is believed that the gene that causes sickle-cell disease also provides natural resistance to malaria, an often-fatal disease. The parts of Africa where malaria is most prevalent, such as Ghana and Nigeria, are also the areas where the incidence of sickle-cell anemia and sickle-cell trait is greatest. It is believed that the gene that causes the sickle-cell trait also gives these people some advantage in surviving malaria. This allows them to live and reproduce, thereby passing the gene along to later generations. However, people with sickle-cell disease (two copies of the HbS sickle-cell gene) do not have this advantage and are more likely to die of malaria.
Resources
Organizations
The NIH National Heart, Lung, and Blood Institute Information Center,
P.O. Box 30105, Bethesda, MD 20824-0105. Posts information about
sickle-cell disease on its website.
Telephone 301-592-8573
http://www.nhlbi.nih.gov/health/public/blood/sickle/sca_fact.txt
Sickle-Cell Disease Association of America, Inc., 200 Corporate Pointe,
Suite 495, Culver City, CA 90230-7633. This association has a hotline
staffed by counselors and provides free education materials.
Telephone 800-421-8453